Статика и термодинамика адсорбции полимеров. Термодинамика адсорбции
Кузнецова Е.С. и Буряк А.К. провели сопоставление термодинамических характеристик адсорбции аминокислот и их ассоциатов. В работе было исследовано влияние строения аминокислот, их димеров и ассоциатов с компонентами элюента на их адсорбцию на поверхности углеродных материалов. Проведен молекулярно-статистический расчет термодинамических характеристик адсорбции (ТХА) для ароматических аминокислот (фенилаланина, тирозина), гетероциклической аминокислоты (триптофана) и их димеров с трифторуксусной кислотой (ТФУ) на поверхности графитированной термической сажи (ГТС). Полученные данные сопоставлены с закономерностями удерживания аминокислот на пористом графитированном углероде Гиперкарбе в условиях обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФ ВЭЖХ). Показано, что ТХА и величины удерживания аминокислот возрастают с увеличением углеродной цепи этих соединений.
Школиным А. В., и Фомкиным А. А. был проведен анализ поведения термодинамических функций (дифференциальной мольной изостерической теплоты адсорбции, энтропии, энтальпии и теплоемкости) адсорбционной системы метан-микропористый углеродный адсорбент АУК в зависимости от параметров адсорбционного равновесия в интервалах температур от 177.65 до 393 К и давлений от 1 Па до 6 МПа. Учет влияния неидеальности газовой фазы и неинертности адсорбента привел к появлению температурной зависимости изостерической теплоты адсорбции, особенно в области высоких давлений адсорбтива. Для исследованной системы основное влияние на термодинамические функции адсорбционной системы оказывает неидеальность газовой фазы. Поправка на неинертность адсорбента в данном интервале параметров адсорбционной системы составляет не более 2.5%.
В институте общей и неорганической химии Академии наук Республики Узбекистан Муминов С.З. в своей работе исследовал изменения поверхностных свойств и пористой структуры монтмориллонита при замещении обменных катионов минерала на полигидроксиалюминиевые. Предварительное термовакуумирование оказывает существенное влияние на адсорбционные свойства полигидроксиалюминиевого монтмориллонита по отношению к метиловому спирту. По данным серий изостер адсорбции СН3 на дегидратированных натриевом и модифицированном монтмориллонитах, измеренных в широком температурном интервале, установлены зависимости теплоты адсорбции от количества адсорбированного вещества.
Н.С. Казбанов, А.В. Матвеева и О.К. Красильникова проведели исследование адсорбции фенола из водных растворов активированными углями типа ФАС, ПАУ и углеродным войлоком при температурах 293, 313 и 343К в интервале концентраций 5 - 250 ммоль/л. Серию образцов последовательно активированного угля ФАС, отличающегося узким распределением пор по размерам, получали карбонизацией полимеров на основе фурфурола. ПАУ -это микропористый полимерный активированный уголь. Углеродный войлок представляет собой волокнистый материал на основе гидратцеллюлозных волокон. Параметры пористой структуры адсорбентов определяли по изотермам адсорбции паров азота при 77 К (ASAP-2020, Micromeritics, USA). Исследования адсорбции растворов проводили ампульным методом в термостате. Отобранные пробы анализировали методом спектрофотомерии. Анализ полученных изотерм жидкофазной адсорбции был проведен с помощью теории объёмного заполнения микропор (ТОЗМ) по уравнению Дубинина- Радушкевича (ДР).
Влияние температуры на сорбцию из жидких растворов неоднозначно. С одной стороны, для микропористых адсорбентов проникновение молекул в поры, сравнимые по размеру с этими молекулами, зависит от кинетической энергии и, соответственно, увеличивается с температурой. С другой стороны, физическая адсорбция представляет собой экзотермический процесс, и адсорбция уменьшается с температурой. Соотношением этих факторов для каждой системы и определяется ход температурной зависимости адсорбции.
Уникальность системы адсорбент - фенол состоит в том, что она имеет обратную температурную зависимость изотерм адсорбции т.к. при увеличении температуры от 293 до 313 К предельная величина адсорбции растёт, что по-видимому связано с молекулярно-ситовым эффектом: с увеличением температуры молекулы фенола способны проникать в более узкие поры углеродных материалов. Адсорбция происходит в основном в микропорах, поскольку адсорбенты обладают небольшим количеством мезопор. По мере увеличения размера микропор величины предельной адсорбции значительно увеличиваются, достигая 2,9 ммоль/г для ПАУ, 8,5 ммоль/г для ФАС и 12,7 ммоль/г для войлока. Полученные изотермы адсорбции хорошо описываются уравнением ДР с показателем степени, равным 2.
Текущая страница: 6 (всего у книги 19 страниц) [доступный отрывок для чтения: 13 страниц]
Шрифт:
100% +
34. Природа адсорбционных сил
Взаимодействие между молекулами адсорбтива с поверхностью адсорбента при т. н. физической адсорбции может быть обусловлена различными причинами. Тогда потенциал, который обусловливает взаимодействие одной молекулы адсорбента с одним атомом неполярного адсорбтива, можно выразить так:
θ = − Сr 6 + Br 12 ,
где r – расстояние между центрами частиц; С – константа дисперсионного притяжения; В – константа, которая характеризует энергию сил отталкивания.
Совершенно очевидно, что на сравнительно отдаленных расстояниях должны преобладать силы притяжения, а на расстояниях близких – силы отталкивания. Также на определенных расстояниях эти силы должны быть равными, что будет соответствовать минимуму свободной энергии. Но важно отметить, что при адсорбции дисперсионные силы действуют одновременно между каждой неполярной частицей.
Поскольку энергия взаимодействия частиц может быстро убывать с расстоянием, то для определения потенциала адсорбционных сил достаточно провести суммирование на ближайших атомах адсорбента. Важным является то, что при адсорбции сложных неполярных молекул потенциальную энергию можно приближенно подсчитать как сумму всех потенциальных энергий адсорбции звеньев молекулы.
Если же адсорбент состоит из ионов, то к действию уже известных дисперсионных сил может прибавляться действие индукционных сил притяжения диполей которые индуцированы в молекулах адсорбтива электрическим полем, которое, в свою очередь, создается ионами решетки адсорбента.
При таком взаимодействии доля индукционных сил в адсорбционном взаимодействии может быть пропорциональна поляризуемости молекулы адсорбтива и квадрату напряженности поля на этой поверхности адсорбента.
Если же на полярном адсорбенте происходит адсорбция полярных молекул адсорбтива, то диполи в этом случае поляризуют атомы адсорбента, т. е. как бы индуцируют в них электрические моменты. Вследствие такого влияния индукционное взаимодействие добавляется к дисперсионному.
Само индукционное взаимодействие обычно мало и в зависимости от диполя молекулы адсорбтива и поляризуемости адсорбента может достигать больших значений. В случае, если молекулы адсорбируются на адсорбенте, который имеет на поверхности ионы или диполи, возникает т. н. взаимодействие ионов или диполей адсорбтива с электростатическим полем самого адсорбента.
При этом молекулы адсорбтива могут даже ориентироваться в поле адсорбента, при этом происходит ориентационное кулоновское взаимодействие. Обычно бывает, что энергии индукционного и ориентационного взаимодействия меньше энергии дисперсионного взаимодействия, и поэтому принимается, что энергия межмолекулярного притяжения определяется энергией дисперсионного притяжения.
Также причиной адсорбции может служить образование водородной связи. Связь такого типа может возникать при адсорбции на адсорбентах, которые содержат на поверхности гидроксильные группы таких молекул, как молекулы воды, спиртов, аммиака и аминов. При образовании водородной связи энергия взаимодействия адсорбтива с адсорбентом может быть довольно большой, и теплота, которая выделяется при такой адсорбции, значительно больше теплоты адсорбции веществ, которые сходны по форме и размеру молекул, но не образуют водородной связи.
Важно отметить, что, зная термодинамическое описание поверхностного слоя на границе «адсорбент – адсорбтив», его строение, природу различных видов сил, динамику процесса, можно переходить к изучению более сложных процессов адсорбции.
35. Адсорбция как самопроизвольное концентрирование на поверхности раздела фаз веществ, снижающих межфазное натяжение
Поверхностно-активные вещества делятся на две большие группы: активные и инактивные вещества.
Поверхностно-активные вещества способны накапливаться в поверхностном слое, и при этом происходит положительная адсорбция Г > 0.
Такие виды веществ должны обладать поверхностным натяжением, которое, в свою очередь, должно быть меньше поверхностного натяжения растворителя, или в противном случае накопление вещества в поверхностном слое будет невыгодно, и должны обладать сравнительно малой растворимостью. При достаточно хорошей растворимостью молекулы поверхностно-активных веществ стремятся уйти с поверхности в глубь раствора. Следовательно, поверхностно-активные вещества будут преимущественно выталкиваться из объема жидкости на поверхность.
Но при накоплении веществ на границе раствора в молекулах этих веществ, которые слабо взаимодействуют друг с другом, межмолекулярное взаимодействие в поверхностном слое будет уменьшаться, а поверхностное натяжение будет падать.
Поверхностно-активными веществами относительно водного слоя являются многие виды органических соединений, жирные кислоты с достаточно большим углеводородным радикалом, соли этих кислот (мыла), сульфокислоты и их соли, а также различные виды спиртов и аминов. Характерной особенностью большинства молекул является их дифильность: молекула состоит из двух частей полярной группы и неполярного углеводородного радикала. Обладающая значительным дипольным моментом и хорошо гидратирующая полярная группа может обусловливать сродство поверхностно-активного вещества к водной среде. Но углеводородный радикал является причиной, которая понижает растворимость этих соединений.
Поверхностно-инактивные вещества ПАВ – эти виды вещества, стремящиеся уйти с поверхности жидкости в ее объем, в результате происходит т. н. отрицательная адсорбция Г < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Поверхностно-инактивными веществами в отношении воды являются многие неорганические электролиты: кислоты, щелочи, соли. Молекулы поверхностно-инактивных веществ не имеют гидрофобной части и могут распадаться в воде на хорошо гидратирующие ионы.
Примерами поверхностно-инактивных веществ являются и некоторые органические соединения, у которых неполярная часть молекулы отсутствует или очень мала. К таким веществам можно отнести муравьиную, аминоуксусную кислоты.
В неводных растворителях неорганические электролиты также способны повышать поверхностное натяжение, причем это зависит от растворителя.
Например , при введении иодида натрия в метанол сильно повышается поверхностное натяжение, для этанола поверхностное натяжение больше примерно в 2 раза. Поверхностная активность веществ может зависеть не только от природы вещества, но также от свойств растворителя. Если какой-либо растворитель обладает большим поверхностным натяжением, то данное растворенное вещество может проявлять значительную поверхностную активность.
36. Теории адсорбции
Рассмотрим наиболее распространенные теории адсорбции, описывающие отдельные виды адсорбции на поверхности раздела «твердое тело – газ» или «твердое тело – раствор».
Теория мономолекулярной адсорбции И. Ленгмюра.
1. Адсорбция является локализованной и вызывается силами, близкими к химическим.
2. Адсорбция происходит только на активных центрах – выступах или впадинах на поверхности адсорбента, характеризующихся наличием свободных валентностей. Активные центры считаются независимыми и тождественными.
3. Каждый активный центр способен взаимодействовать только с одной молекулой адсорбата; на поверхности может образоваться только один слой адсорбированных молекул.
4. Процесс адсорбции является обратимым и равновесным; адсорбированная молекула удерживается активным центром некоторое время, после чего десорбируется; через некоторое время устанавливается динамическое равновесие.
Максимально возможная величина адсорбции Г о достигается при условии, что все активные центры заняты молекулами адсорбата. Уравнение изотермы мономолекулярной адсорбции, связывающее величину адсорбции Г с концентрацией адсорбата С , имеет вид:
где b – постоянная для данной пары «адсорбент – адсорбат» величина (отношение констант скоростей десорбции и адсорбции), численно равная концентрации адсорбата, при которой занята половина активных центров.

График изотермы адсорбции Ленгмюра приведен на рисунке 2. Константу b определим графически, проведя касательную к изотерме адсорбции в точке С = 0. При описании процесса адсорбции газов в уравнении концентрация может быть заменена пропорциональной величиной парциального давления. Теория мономолекулярной адсорбции И. Ленгмюра применима для описания процессов адсорбции газов и растворенных веществ при небольших давлениях (концентрациях) адсорбата.
Теория полимолекулярной адсорбции Поляни описывает s-образные изотермы адсорбции, форма которых свидетельствует о возможном взаимодействии адсорбированных молекул с адсорбатом.
1. Адсорбция вызвана физическими силами.
2. Поверхность адсорбента однородна, нет активных центров; адсорбционные силы образуют непрерывное силовое поле вблизи поверхности адсорбента.
3. Адсорбционные силы действуют на расстоянии, большем размера молекулы адсорбата, т. е. у поверхности адсорбента существует некоторый адсорбционный объем, который при адсорбции заполняется молекулами адсорбата.
4. Притяжение молекулы адсорбата поверхностью адсорбента не зависит от наличия в адсорбционном объеме других молекул, вследствие чего возможна полимолекулярная адсорбция.
5. Адсорбционные силы не зависят от температуры, и, следовательно, с изменением температуры адсорбционный объем не меняется.
Уравнение Фрейндлиха. Поверхность адсорбента неоднородна, между адсорбированными частицами происходит взаимодействие, активные центры не являются полностью независимыми друг от друга. Г. Фрейндлих предположил, что число молей адсорбированного газа или растворенного вещества, приходящееся на единицу массы адсорбента (т. н. удельная адсорбция х /m ), должно быть пропорционально равновесному давлению (для газа) или равновесной концентрации (для веществ, адсорбируемых из раствора) адсорбента, возведенной в некоторую степень, которая всегда меньше единицы:
x / m = aP n ; x / m = aC n .
Показатели степени n и коэффициент пропорциональности а определяются экспериментально.
37. Термодинамика процесса адсорбции. Уравнение адсорбции Гиббса
Для изучения явления адсорбции на границе «раствор – газ» нужно установить связь между избытком адсорбированного вещества в слое на поверхности (Г ), концентрацией ПАВ в растворе (с ) и поверхностным натяжением (σ ) на границе раздела фаз «раствор – газ». Целесообразнее рассматривать явления с термодинамических позиций и связывать адсорбцию растворенного вещества с изменением свободной энергии поверхности или ее поверхностного натяжения. Эту связь вывел В. Гиббс в 1876 г, которая получила название «уравнение адсорбции Гиббса» :
Г = – с / RT x dσ / dc .
Еще можно представить уравнение Гиббса, основанное на термодинамике, с использованием изобарно-изотермического потенциала G , химических потенциалов μ 1 и μ 2 , а также с использованием n 1 и n 2 числом молей компонентов. Проанализировав его с учетом энтропии S , объема V и давления P , можно записать следующее уравнение:
dG = – SdT + VdP + σds + μ 1 d n 1 +μ 2 dn 2 .
Приравняем его к нулю, и с учетом постоянной температуры и давления оно упрощается в уравнение вида:
sdσ + n 1 dμ 1 + n 2 dμ 1 = 0.
С учетом того, что для разбавленных растворов химический потенциал второго компонента выражается так:
μ 2 = μ 2 0 + RT lnc ,
а с учетом того, что температура постоянна
dμ 2 = RTdnc,
подставляя это уравнение в
![]()
получаем искомое уравнение адсорбции Гиббса. Исходя из уравнения, можно заметить, что если поверхностное натяжение σ увеличивается с концентрацией с , то концентрация растворенного вещества на поверхностном слое меньше, чем в объеме раствора (т. н. отрицательная адсорбция), и если поверхностное натяжение σ уменьшается с увеличением концентрации с , тогда концентрация в слое больше, чем в объеме (оположительная адсорбция), и, наконец, если σ не зависит от с , то концентрация вещества в слое на поверхности и в объеме одинакова. Уравнение Гиббса было выведено с использованием термодинамики. Практически проверить это уравнение сложно, что связано со сложностью определения концентрации растворенного вещества в слоена поверхности. Опытным путем Б. Мак-Бен установил, что с поверхности раствора с помощью прибора срезался очень тонкий слой жидкости. Дальнейшее определение всех параметров уравнения Гиббса показало, что экспериментально найденные значения адсорбции в пределах ошибки опыта совпадали со значениями, которые вычисляли по уравнению Гиббса. Из-за однородности и гладкости поверхности всякой жидкости при изучении адсорбции на ее поверхности совершенно неприложимы обычные представления об активных центрах. При критической температуре исчезает различие между граничащими фазами, поверхностное натяжение, как правило, становится равным нулю. Адсорбция газов и паров имеет настолько большое практическое применение, что в литературе, особенно в технической, можно встретить это понятие, которое применяют только по отношению к процессам на поверхности твердых тел.
Это понятие, как и наиболее общие закономерности адсорбции, как рассмотренное уравнение Гиббса, применимо ко всем границам раздела фаз. Пользуясь уравнением Гиббса и всеми вытекающими из него положениями, определив величину Г, можно построить изотерму адсорбции.
38. Особенности адсорбции на микропористых материалах. Потенциальная теория Поляни. Адсорбционный потенциал
Теория Поляни рассматривает нелокализованную физическую адсорбцию, которая непосредственно обусловлена вандерваальсовыми силами между адсорбентом и адсорбатом (это можно считать первым положением). Вторым положением этой теории является представление о силовом, (или потенциальном) поле адсорбента, которое распространяется на значительное расстояние от поверхности; слой адсорбции, который возникает в этом поле, полимолекулярен. Если рассматривать адсорбцию газов, тогда плотность этого слоя убывает по определенной нормали от поверхности. Если рассматривать адсорбцию паров, тогда на поверхности образуется жидкий слой определенной толщины. Поле в теории Поляни рассматривают как ряд эквипотенциальных поверхностей, каждая поверхность соответствует определенному значению потенциала ε , причем каждая последующая поверхность будет меньше, чем предыдущая. Каждая такая поверхность в пространстве вырезает слои определенного объема, обозначенного как v i . Задачей теории Поляни является нахождение перехода от обычных координат изотермы (x, p ) к параметрам поля ε i и v i , с дальнейшим установлением связи между этими основными параметрами. Первая часть задачи, которую заложил Поляни, достаточно сложна, и во множестве случаев не может иметь определенных решений, но для случая адсорбции паров эта часть задачи решается в первом приближении очень просто. Для жидкого адсорбционного слоя заполненная часть объема будет равна:
v i = х(М/d) ,
где d – плотность вещества в жидком состоянии.
В своей теории M. Поляни вводит еще одно положение об отсутствии т. н. экранирования поля в процессе адсорбции, величина ε в данной теории пространства является величиной постоянной (что-то наподобие гравитационного потенциала) независимо от того, существуют ли определенные молекулы адсорбата между данной точкой и твердой поверхностью или же все пространство является свободным. Поляни вводит понятие адсорбционного потенциала ε , который представляет собой изотермическую работу сжатия пара при переводе его от равновесного давления р в объемной фазе вдали от поверхности в область поверхностного слоя с давлением насыщенного пара р 0 тогда выражение для определения потенциала будет иметь вид:
ε = RT lnр 0 / р .
При помощи такого уравнения можно перейти от координат x, p к координатам ε и v и получить кривую, которая получила название «характеристическая». Поляни в своих опытах обнаружил, что такие кривые, построенные по экспериментальным данным полученных изотерм, обладают таким свойством: они инвариантны по отношению к Т, или, говоря иначе, все кривые такого типа могут ложиться на одну кривую ε −ε .
Такое положение М. Поляни принял в качестве постулата, т. е.:
Указанное свойство Поляни имеет огромное практическое значение, оно может по одной экспериментальной изотерме адсорбции построить семейство изотерм.
Теория Поляни не дает аналитического выражения для изотермы или функции потенциала от объема, но позволяет вычислить координату для любой заданной температуры, если известна хотя бы одна изотерма. Такой результат очень важен для технологических расчетов, потому что для сходных газов на одном адсорбенте кривые адсорбции могут оказаться близкими друг к другу и могут быть во многих случаях совмещены.
39. Характеристическая кривая адсорбции. Температурная инвариантность и аффинность характеристических кривых
Силовое поле, которое возникает у поверхности адсорбента, во многом может быть схоже с гравитационным полем. В адсорбционном поле можно представить потенциальные поверхности, т. е. поверхности для которых характерен один и тот же адсорбционный потенциал. Под понятием адсорбционного потенциала θ следует понимать не что иное, как работу, совершаемую против сил адсорбции при перемещении 1 моля адсорбтива из определенной точки поля в некоторую газовую фазу. Максимальный адсорбционный потенциал будет существовать на границе «адсорбент – адсорбционный объем». Но на границе «объем – газовая фаза» (именно там кончается действие адсорбционных сил) потенциал адсорбции должен быть равен нулевому значению. Изменение адсорбционного потенциала при изменении адсорбционного объема можно представить в виде кривых. Впервые это сделал М. Поляни. Подобные типы кривых не зависят от температуры и могут быть характерны для каждого конкретного адсорбента, такие типы кривых принято называть характеристическими кривыми адсорбции. Теория полимолекулярной адсорбции принимает, что для объема адсорбции применимо уравнение состояния газа. Следовательно, изотермы, которые характеризуют зависимость плотности адсорбтива от объема для разной температуры, напоминают изотермы зависимости давления от объема. При низкой температуре силы адсорбции на поверхности могут вызвать конденсацию пара в жидкость определенной плотности. При температурах более низких, чем критическая, при конденсации весь адсорбционный объем будет заполнен жидкостью. В этом случае кривая адсорбции будет идти почти параллельно оси абсцисс, которая связана с малой сжимаемостью жидкости. Затем кривая адсорбции на границе «объем – газовая фаза» резко опускается вниз, и, соответственно, плотность адсорбтива достигает значения некоторой плотности газовой фазы. При температурах более высоких, чем критическая, адсорбтив может вести себя как идеальный газ, и график будет выражаться как изотерма зависимости для идеального газа при условии, что pV = RT . При таких условиях адсорбированный газ будет иметь максимальную плотность у самой поверхности адсорбента и иметь минимальную при непосредственной близости от газовой фазы. Причем в этом случае важно отметить, что плотность адсорбтива в адсорбционном слое нигде не достигает плотности самой жидкости. И если температура очень близка к критической, зависимость плотности от объема будет выражаться кривой, близкой по виду к изотерме, которая описывается уравнением Ван-дер-Ваальса. При таком раскладе часть адсорбированного вещества будет находиться в адсорбированном объеме в жидком состоянии, а часть адсорбированного вещества – в газообразном. Тогда кривая будет наиболее резко снижаться в участке, который отвечает переходу от жидкости к газу. Если построить характеристическую кривую по опытной изотерме адсорбции одного из адсорбтивов, а зная соответствующие коэффициенты аффинности для какого-нибудь другого адсорбтива, можно найти изотерму адсорбции и построить ее для другого адсорбтива. Потенциальная теория адсорбции дает возможность вычислить различные изотермы адсорбции различных паров на одном и том же адсорбенте, причем по характеристической кривой, которая получена из изотермы адсорбции одного пара, т. к. соотношение адсорбционного потенциала не зависит от адсорбционных объемов.
Аффинность (от лат. affinis – «родственный») – хроматография по сродству. Метод очистки и разделения белков основан на их избирательном взаимодействии с лигандом, ковалентно связанным с инертным носителем (аффинная хроматография). Измерение аффинности токсиканта к рецептору, по сути, представляет собой экспериментальное изучение зависимости между количеством вещества, добавляемого в инкубационную среду, и количеством образующегося в результате взаимодействия токсикант-рецепторного комплекса.
Адсорбция как самопроизвольное концентрирование молекул на поверхности сопровождается понижением энтропии системы. Так как критерием самопроизвольности процесса является
∆Н - T· ∆S = ∆G< 0,
то адсорбция возможна только при ∆Н < 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. При повышении температуры равновесие смещается в сторону эндотермического процесса, т. е. десорбции.
Адсорбция на поверхности твердого тела
1. Мономолекулярная адсорбция.
По теории Ленгмюра молекулы адсорбтива взаимодействуют с поверхностью адсорбента, образуя в итоге мономолекулярный слой. B этом случае степень заполнения () поверхности адсорбируемым веществом при адсорбции из газовой фазы
из жидкости
где К - константа равновесия (константа адсорбции);
р - парциальное давление адсорбируемого газа;
с - концентрация адсорбируемого вещества.
Зависимость β от р (или с) представлена графиком (изотерма адсорбции, Т = const) на рис. 1.3.

Рис. 1.3. Степень заполнения поверхности адсорбируемым веществом
При малых концентрациях и парциальных давлениях адсорбция пропорциональна концентрации или парциальному давлению:
р<< 1, β ≈ К· р илис<< 1, β ≈ К· с, т.е. начальный участок изотермы приблизительно линеен, причем tg α = К(tg α определяют по наклону кривой при р (или с) → 0: или ).
Если - количество молей адсорбированного вещества на 1 г адсорбента; - максимально возможное количество молей адсорбированного вещества на 1 г адсорбента ("емкость монослоя"), то
Подставляя β в уравнение (1.3) (для случая адсорбции из газовой фазы концентрацию с в уравнениях следует заменить на давление р ), получаем:
 (1.6)
(1.6)
Так как и К в данной паре адсорбент-адсорбтив являются константами (при T =const), то по зависимости можно найти и К (рис. 1.4).

Рис. 1.4. Графическое решение уравнения адсорбции
получают путем экстраполяции экспериментальной линейной зависимости к () = 0; и, так как , то , .
Величину можно использовать для определения удельной поверхности адсорбента УД (в м 2 на 1 г адсорбента), если известна площадь ω, занимаемая на поверхности одной молекулой адсорбтива (определяется из размеров молекулы):
УД = · ω · Nа, (1.7)
где Nа - число Авогадро (Nа = 6,02 · 10 23).
В свою очередь, известную величину УД можно использовать для расчета или ωлюбого вещества по его адсорбции на данном адсорбенте.
2. Полимолекулярная адсорбция.
Уравнение (1.5) описывает кривую с насыщением, т.е. при
р (или с) → ∞ стремится к предельному значению, равному (рис. 1.5,а).

Рис.1.5. Изотермы адсорбции:
а – адсорбция с насыщением; б – полимолекулярная адсорбция
Однако в некоторых случаях изотермы адсорбции выглядят как показано на рис. 1.5,б, т.е. не достигает предела даже при высоких р (или с).
Зависимости типа показанной на рис. 1.5,б соответствуют полимолекулярной адсорбции. Как правило, такие изотермы характерны для веществ с сильными межмолекулярными взаимодействиями (например, для воды). Когда центры адсорбции на поверхности адсорбента заняты (мономолекулярный слой насыщен), "посадка" следующих молекул адсорбата происходит за счет межмолекулярных взаимодействий с уже адсорбированными молекулами (рис.1.6). Теплота такой адсорбции близка по абсолютной величине, но противоположна по знаку теплоте испарения соответствующей жидкости (подумайте, почему).

Рис.1.6. Схема адсорбции:
а - мономолекулярная адсорбция; б - полимолекулярная адсорбция
По мере приближения р к давлению насыщенного пара адсорбируемого вещества оно начинает конденсироваться на поверхности адсорбента, в результате быстро растет с ростом р .
Основные определения и способы классификации адсорбционных процессов.
Адсорбция относится к явлениям, происходящим вследствие самопроизвольного уменьшения поверхностной энергии.
Адсорбция – процесс самопроизвольного обратимого или необратимого перераспределения компонентов гетерогенной системы между поверхностным слоем и объемом гомогенной фазы.
В многокомпонентных системах в поверхностный слой предпочтительнее переходит компонент, который сильнее снижает межфазное натяжение. В однокомпонентных системах при формировании поверхностного слоя происходит изменение его структуры (определенная ориентация атомов и молекул, поляризация), называемое автоадсорбцией .
Более плотную фазу, на которой локализованы адсорбционные взаимодействия называют адсорбентом . Вещество, перераспределяемое между объемом гомогенной фазы и поверхностным слоем, обозначают термином «адсорбат ».
В ряде случаев процесс адсорбции является обратимым. В этом случае при определенных условиях часть адсорбированных молекул в результате молекулярно-кинетических явлений может перейти из поверхностного слоя в объем фазы. Процесс, обратный адсорбции, называют десорбцией .
Способы классификации адсорбционных процессов.
Классификация адсорбционных процессов по агрегатному состоянию взаимодействующих фаз. В зависимости от агрегатного состояния смежных фаз различают следующие типы адсорбционных процессов:
Адсорбция газов на твердых адсорбентах;
Адсорбция растворенных веществ на границах раздела «твердое тело – жидкость» и «жидкость – жидкость»;
Адсорбция поверхностно-активных веществ на границе раздела «жидкость – газ».
Классификация адсорбционных процессов по механизму взаимодействия адсорбента и адсорбата. Адсорбцию можно рассматривать как взаимодействие молекул адсорбата с активными центрами адсорбента. По механизму их взаимодействия подразделяют следующие виды адсорбции:
1) физическая (молекулярная) адсорбция – взаимодействие между молекулами адсорбата и адсорбента осуществляется за счет сил Ван-дер-Ваальса, водородных связей (без протекания химических реакций);
2) химическая адсорбция (хемосорбция) – присоединение молекул адсорбата к активным центрам адсорбента происходит в результате протекания химических реакций различных типов (за исключением реакций ионного обмена);
3) ионообменная адсорбция (ионный обмен) – перераспределение вещества адсорбата между раствором и твердой фазой (ионитом) по механизму реакций ионного обмена.
Для количественного описания адсорбционных процессов применяют две величины.
1) Абсолютная адсорбция – количество (моль) или масса (кг) адсорбата на единицу площади поверхности или массы адсорбента. Обозначение – А; размерность: моль/м 2 , моль/кг, кг/ м 2 , кг/кг.
2) Гиббсовская (избыточная) адсорбция – избыток вещества адсорбата в поверхностном слое определенной толщины по сравнению с его количеством в объеме гомогенной фазы, отнесенный к единице площади поверхности или массы адсорбента. Обозначение – Г; размерность: моль/м 2 , моль/кг.
Связь между абсолютной и избыточной адсорбции можно проиллюстрировать с помощью уравнения:
Г = А – с * h (3.1)
где с – равновесная концентрация вещества в объеме фазы, моль/м 3 ;
h - толщина поверхностного слоя, условно принимаемая равной 10 -9 м.
В многокомпонентных гетерогенных системах при перераспределении того или иного компонента между объемом гомогенной фазы и поверхностным слоем справедливо уравнение для избыточной внутренней энергии поверхности:
U = T * S + s * s + Sm i * n i (3.2)
Приведя все члены уравнения к единице площади межфазной поверхности, получим:
U s = T * S s + s + Sm i * Г i (3.3)
где Г i = n i / s – избыток i -го компонента в поверхностном слое, то есть гиббсовская адсорбция.
Для однокомпонентной системы уравнение (3.3) примет вид:
G s = s + m * Г (3.4)
где G s = U s - T * S s – энергия Гиббса поверхности или работа создания единицы площади поверхности;
m * Г – уплотнение вещества адсорбируемого вещества в поверхностном слое.
Исходя из уравнения (3.4) можно сделать вывод о том, что при адсорбции работа по созданию межфазной поверхности складывается из работы образования поверхности (разрыва когезионных связей в объеме фазы адсорбата) и уплотнения вещества в поверхностном слое.
В состоянии динамического равновесия между адсорбентом и адсорбатом изменение энергии Гиббса гетерогенной системы ΔG = 0, термодинамика процесса адсорбции описывается уравнением, получившим название фундаментальное адсорбционное уравнение Гиббса :
Ds = SГ i * dm i (3.5)
Данное уравнение является универсальным, так как справедливо для всех типов адсорбционных процессов
Частные случаи адсорбционного уравнения Гиббса.
1) Адсорбция из растворов.
Для химического потенциала i -го компонента системы при протекании адсорбции на границах раздела «жидкость – твердый адсорбент» и «жидкость – газ» справедливы уравнения:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R*T* d ln a i (3.7)
где m i 0 - химический потенциал i -го компонента системы при стандартных условиях;
a i – активность i -го компонента системы при стандартных условиях.
Исходя из этого, адсорбционное уравнение Гиббса примет вид:
Г i = - a i / R*T * (ds / da i) (3.8)
Для растворов неэлектролитов принимаем a i = с i , тогда:
Г i = - с / R*T * (ds / dс) (3.9)
Для растворов электролитов:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
где с ± - средняя ионная концентрация раствора;
n - стехиометрический коэффициент.
2) Адсорбция веществ из газовой фазы.
В соответствии с уравнением Менделеева-Клайперона:
Р = с * R*T (3.11)
В связи с этим, уравнение Гиббса для адсорбции газов на твердых адсорбентах записывают в следующей форме:
Г i = - Р / R*T * (ds / dР) (3.12)
На практике адсорбционное уравнение Гиббса позволяет по данным измерения поверхностного натяжения при различных значениях концентрации жидкости или равновесного давления газа рассчитать величину адсорбции веществ в межфазном слое, для которого определено поверхностное натяжение.
В случае взаимодействия двух атомов:
U – энергия взаимодействия;
U = U ПРИТЯЖ. + U ОТТАЛК.
 - уравнение
Леннарда-Джонса
,
c,
b,
m
= const
- уравнение
Леннарда-Джонса
,
c,
b,
m
= const
В случаях взаимодействия атомов с твердой поверхностью, необходимо провести суммирование всех взаимодействий.
х– расстояние до поверхности
r – радиус действия сил притяжения
dV – объем
n – число молекул поверхности
U АДС. – энергия адсорбционного взаимодействия



В случае адсорбции притяжение усиливается. И в случае при взаимодействии типа неполярное-неполярное адсорбция преимущественно локализуется в углублениях.
Электростатическое взаимодействие.
Полярный адсорбент – неполярный адсорбат
Неполярный адсорбент – полярный адсорбат
Полярный адсорбент – полярный адсорбат.
М олекулу
адсорбата представляют как диполь, а
адсорбента – как проводник, в котором
молекула адсорбата индуцирует диполь
зеркально симметрично по отношению к
данному.
олекулу
адсорбата представляют как диполь, а
адсорбента – как проводник, в котором
молекула адсорбата индуцирует диполь
зеркально симметрично по отношению к
данному.
X – расстояние до середины
При взаимодействии возникает потенциал:
 ,
,
 - дипольный момент.
- дипольный момент.
Потенциал стремится принять максимальное значение, т.е. диполи стремятся сориентироваться перпендикулярно к поверхности.
Поскольку повышение температуры способствует росту Броуновского движения, оно приводит к торможению процесса адсорбции.
В случае электростатического взаимодействия адсорбат преимущественно локализуется на выступах.
Фундаментальное адсорбционное уравнение.
В случае адсорбции происходит перераспределение компонента, а значит, изменяется химический потенциал. Процесс адсорбции можно рассматривать как переход поверхностной энергии в химическую.
Объем слоя = 0, тогда обобщенное уравнение I и II закона термодинамики:
T
= const;
(1) = (2) =>


Для двухкомпонентной системы:
,
 ,
,
 =>
=>
=>
 - адсорбционное
уравнение Гиббса
.
- адсорбционное
уравнение Гиббса
.
Для случая адсорбции
тв. тело – газ:
,
 ,
,

 - изотерма
- изотерма
 - изобара
- изобара
 - изопикна
- изопикна
 - изостера
- изостера
Изотерма, изопикна, изостера связаны друг с другом.
Т.к. адсорбция
функция




Изотерма Генри Изотерма Лангмюра



Термодинамика. Адсорбция.
Для конденсированных сред:
 ,
,
 ,
,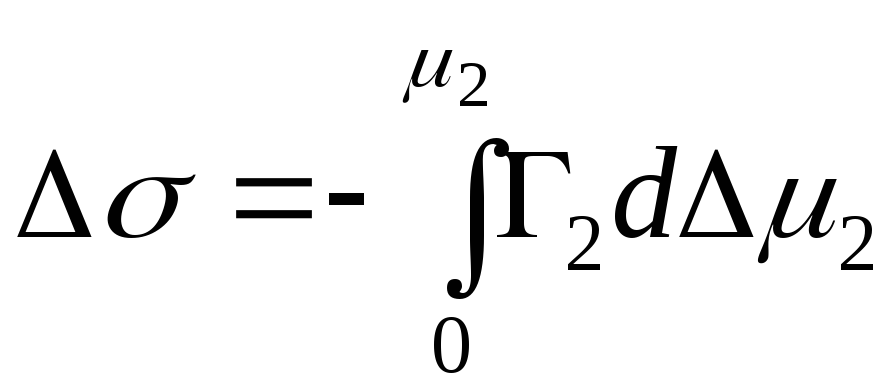
 - интегральное
изменение энергии Гиббса
.
- интегральное
изменение энергии Гиббса
.

P–давление над искривленной поверхностью, Р S –давление над плоской поверхностью
 - адсорбционный
потенциал
- адсорбционный
потенциал
Дифференциальное
изменение энтрапии

 ,
Г = const
,
Г = const
- дифференциальное изменение энтропии
- дифференциальная энтальпия адсорбции
 - изостерическая
теплота адсорбции
- изостерическая
теплота адсорбции
 - теплота
конденсации
- теплота
конденсации
 - чистая
теплота адсорбции
- чистая
теплота адсорбции

 ,
,




Qa
– интегральная теплота адсорбции,

Qra – интегральная чистая теплота адсорбции,

Уравнение Генри
Исследование адсорбции затрудняется неоднородностью поверхности, поэтому простейшие закономерности получают для однородных поверхностей.
Рассмотрим взаимодействие газов с твердой поверхностью, когда осуществляется переход газа из равновесного состояния в объеме в равновесное состояние на поверхности. Этот случай аналогичен равновесию газов в поле силы тяжести.
 ,
,
 ,
=>
,
=> -уравнение
Генри
-уравнение
Генри
 - коэффициент
распределения
- коэффициент
распределения
В процессе адсорбции происходит изменение химических потенциалов.
Для объемной фазы:

Для газа на
поверхности:

В состоянии
равновесия
 ,
т.е.
,
т.е.

В уравнении Генри константа не зависит от концентрации
Уравнение Генри выполняется в области низких давлений и концентраций. По мере роста концентрации возможны 2 типа отклонений от закона Генри:
1 – положительные отклонения, D уменьшается, А уменьшается
2 – отрицательные отклонения, D – возрастает, А – возрастает.
Тип отклонения определяется преобладанием тот или иного типа взаимодействия адсорбент-адсорбат.
При сильном адгезионном взаимодействии коэффициенты активности возрастают – положительное отклонение. В случае когезионных взаимодействий наблюдаются отрицательные отклонения.
Мономолекулрная адсорбция.
Изотерма Лангмюра.
Простейшие закономерности были получены в теории генри. Ленгмюр предложил теорию, согласно которой, адсорбция рассматривается как квазихимическая реакция. При этом:
Поверхность энергетически однородна.
Адсорбция локализована, каждый адсорбционный центр взаимодействует с одной молекулой адсорбата.
Молекулы адсорбата не взаимодействуют друг с другом.
Адсорбция монослойна.

 - поверхность,
- поверхность, - адсорбат,
- адсорбат, - адсорбционный комплекс.
- адсорбционный комплекс.
 ,
тогда концентрация адсорбционных мест:
,
тогда концентрация адсорбционных мест:
 ,
, - предельная адсорбция.
- предельная адсорбция.
 ,
тогда константа реакции:
,
тогда константа реакции:


 - уравнение Лангмюра.
- уравнение Лангмюра.
Зависимость адсорбции от концентрации
1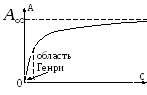 )
)
 ,
,
2) область высоких концентраций
 - предельная
адсорбция, образование мономолекулярного
слоя
- предельная
адсорбция, образование мономолекулярного
слоя
Для энергии Гиббса: .
g – энтропийный множитель.
В случае изотермы
Генри энергия Гиббса характеризует
переход адсорбата из стандартного
состояния в объёме в стандартное
состояние на поверхности. В случае
изотермы Ленгмюра
 характеризует степень сродства адсорбента
и адсорбата.
характеризует степень сродства адсорбента
и адсорбата.
 находят из изобары
Вант-Гоффа.
находят из изобары
Вант-Гоффа.

 ,
тогда
,
тогда
 ,
отсюда
,
отсюда .
.

 - степень заполнения
поверхности.
- степень заполнения
поверхности.




 - число свободных
мест,
- число свободных
мест,
 - число занятых мест.
- число занятых мест.
 ,
,

Т.е. в области высоких концентраций число свободных мест обратно пропорционально количеству адсорбата.
Адсорбция смеси газов на однородной поверхности.
В этом случае процесс адсорбции рассматривают как две параллельно протекающие реакции.
 (1)
(1)

 (2)
(2)


Адсорбция смеси газов на неоднородной поверхности.
В случае неоднородной поверхности нельзя ограничиваться средними заполнениями.
В следствие конкурентной борьбы, на участках различных типов возможна локализация различных адсорбатов.
В этом случае
отношение
 .
.
 ,
,
 - давление насыщенного пара адсорбата.
- давление насыщенного пара адсорбата.
 ,
,
 - теплоты адсорбции.
- теплоты адсорбции.
«+» - симбатная зависимость, «-» - антибатная зависимость, «Н» - корреляции нет.
«+» - адсорбция протекает по одинаковому механизму. На наиболее энергетически выгодных участках преимущественно адсорбируется газ, обладающий большим сродством к поверхности.
«-» - адсорбция протекает по различным механизмам и до определенного момента времени конкурентной борьбы за поверхность нет.
Мономолекулярная адсорбция преимущественно реализуется при физической адсорбции газов при малых значениях p , а также на границе раздела жидкость/газ.
Полимолекулярная адсорбция.
Теория БЭТ (Брунауэр, Эммет, Теллер).
В случае, когда образование монослоя недостаточно для компенсации поверхностной энергии, адсорбция полимолекулярна и её можно рассматривать как результат вынужденной конденсации под действием поверхностных сил.
Основные положения:
При попадании молекулы адсорбата на занятое место образуется кратный комплект.
По мере приближения p к p s уменьшается число свободных адсорбционных мест. Первоначально увеличивается, а затем уменьшается число мест, занятых единичными, двойными и т.д. комплектами.
При p =p s адсорбция переходит в конденсацию.
Горизонтальные взаимодействия отсутствуют.
Для первого слоя выполняется изотерма Лангмюра.
Поверхность рассматривается как совокупность адсорбционных мест. Справедливо условие динамического равновесия: скорость конденсации на свободных местах равна скорости испарения с занятых.
a – коэффициент конденсации (доля молекул, сконденсировавшихся на поверхности);
 ,
,
Zm – максимальное число свободных мест.
 -
частота колебаний атомов в направлении
перпендикулярном к поверхности.
-
частота колебаний атомов в направлении
перпендикулярном к поверхности.
Для первого слоя условия динамического равновесия:

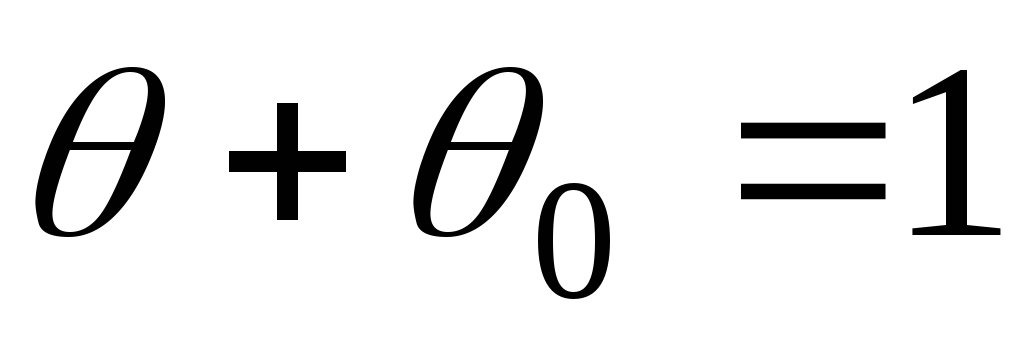

 ,
тогда
,
тогда
 - уравнение Лангмюра.
- уравнение Лангмюра.
Для второго слоя
будет справедливо:

Для i-го
слоя:

Для упрощения принимают, что а и ν одинаковы для всех слоев, кроме первого. Для всех слоев кроме первого теплота адсорбции постоянна. Для последнего слоя теплота адсорбции равна теплоте конденсации. В результате было получено уравнении
 (*)
(*)
C – константа,
В случае теории БЭТ, константа С характеризует энергию Гиббса чистой адсорбции. Уравнение содержит только одну константу, а также это уравнение очень важно для определения удельной поверхности адсорбента.
Поскольку в результате адсорбции теплота выделяется, определение удельных поверхностей ведут при низких температурах.
 ????????????
????????????


Основной недостаток теории – пренебрежение горизонтальными взаимодействиями в пользу вертикальных.
Уравнение выполняется
в области значений
 от 0,05 до 0,3.
от 0,05 до 0,3.
Там, где
 <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 >
0,3 – сказывается взаимодействие адсорбат
– адсорбат.
>
0,3 – сказывается взаимодействие адсорбат
– адсорбат.
Учет взаимодействий адсорбат-адсорбат.
Взаимодействия проявляются при адсорбции на неполярной поверхности разветвленных молекул или молекул. Способных образовывать ассоциаты. В этом случае изменяется форма изотерм адсорбции.
А дсорбент
не полярен.
дсорбент
не полярен.
Графику 1 соответствуют слабые взаимодействия адсорбат-адсорбат, сильное адсорбат-адсорбент.
Графику 2 соответствуют сильное взаимодействие адсорбат-адсорбат, сильное адсорбат-адсорбент.
Графику 3 соответствуют сильное взаимодействие адсорбат-адсорбат, слабое адсорбат-адсорбент.
 ,
,

В случае взаимодействия между молекулами адсорбата необходим учет изменения коэффициентов активности. И это уравнение записывают в виде:
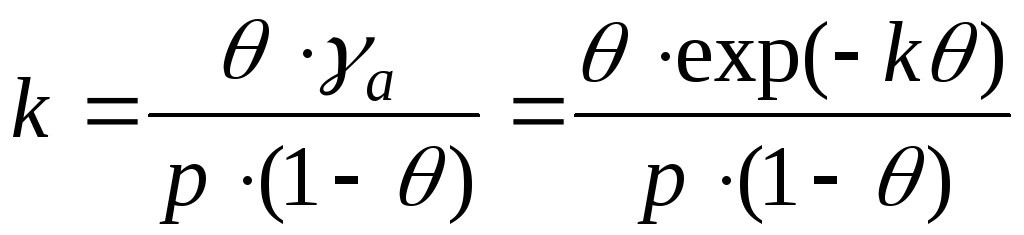 - уравнение Фрункина,
Фаулера, Гугенгейма.
- уравнение Фрункина,
Фаулера, Гугенгейма.
k – аттракционная постоянная.
Потенциальная теория Поляни.
Данная теория не выводит какого-либо типа изотермы адсорбции, а дает возможность расчета изотерм при другой температуре.
Адсорбция – это результат притяжения адсорбата к поверхности адсорбента за счет действия адсорбционного потенциала, который не зависит от присутствия других молекул и зависит от расстояния между поверхностью и молекулой адсорбата.
 ,
,
 - адсорбционный потенциал.
- адсорбционный потенциал.
Поскольку поверхность
неоднородная, расстояние заменяют на
адсорбционный объём
 .Адсорбционный
объём
– это
объём, заключенный между поверхностью
и точкой, соответствующей данному
значению
.Адсорбционный
объём
– это
объём, заключенный между поверхностью
и точкой, соответствующей данному
значению .
.
Адсорбционный потенциал – это работа перенесения 1 моль адсорбата вне данного адсорбционного объёма в данную точку адсорбционного объёма (или работа переноса 1 моль насыщенного пара адсорбата, находящегося в равновесии с жидким адсорбатом в отсутствии адсорбента в равновесную с адсорбентом паровую фазу).

Характеристическая кривая
 - адсорбционный
потенциал,
- адсорбционный
потенциал,

Для данного
адсорбента и различных адсорбатов
справедливо:

Для разных типов
адсорбатов
 ,
,
где
 потенциалы для изотерм адсорбции при
относительных давлениях
потенциалы для изотерм адсорбции при
относительных давлениях для
адсорбата 1 и для адсорбата 2. Это отношение
есть величина постоянная.
для
адсорбата 1 и для адсорбата 2. Это отношение
есть величина постоянная.
 - коэффициент
аффинности
- коэффициент
аффинности
Теория капиллярной конденсации.
Протекание процесса адсорбции во многом зависит от структуры пористого тела.
|
Микропористые | |
|
Переходнопористые | |
|
Макропористые |
В случае микропористых
сорбентов, поля адсорбционных сил
перекрываются. В случае макропористых
сорбентов, поры выполняют роль транспортных
каналов. Процессы коденсации наиболее
значимы в переходнопористых телах.
Капиллярная конденсация начинается
при определенных значениях p
и
 ,
когда часть поверхностной энергии уже
скомпенсирована. Необходимое условие
– поверхность должна быть самчиваема.
Процее описываетсяуравнением
Томпсона – Кельвина
.
,
когда часть поверхностной энергии уже
скомпенсирована. Необходимое условие
– поверхность должна быть самчиваема.
Процее описываетсяуравнением
Томпсона – Кельвина
.

 - для случая
смачивания, центр кривизны находится
в газовой фазе.
- для случая
смачивания, центр кривизны находится
в газовой фазе.
В случае капиллярной конденсации изотерма адсорбции имеет гистерезисный вид. Процессу адсорбции соответствует нижняя ветвь, процессу десорбции – верхняя.
Все виды пор можно свести к трем видам:
|
Конические |
Цилиндрические с одним закрытым концом |
Цилиндрические с двумя открытыми концами |
|
Процессное заполнение осуществляется со дна поры. Изтерма адсорбции и изотерма десорбции в этом случае совпадают, поскольку процесс адсорбции начинается со сферы и процесс десорбции также начинается с исчезновения некоторых сфер.
↓ |
Гистерезиса нет. Прямой и обратный ход описываются уравнением:
|
Дна нигде нет, заполнение поры пойдет по стенкам цилиндра.
цилиндр: Изотерма и будет иметь гистерезисный вид.
↓ |


 - сфера,
- сфера, ,
,



В условиях смачивания конденсация
осуществляется при более низких
давлениях, что энергетически выгодно.
По десорбционной ветви получают кривые
распределения пор по размерам.
условиях смачивания конденсация
осуществляется при более низких
давлениях, что энергетически выгодно.
По десорбционной ветви получают кривые
распределения пор по размерам.
Максимум
дифференциальной кривой смещен влево
относительно точки перегиба интегральной.
Общий объём малых пор невелик, однако
имеет большие значения площади
поверхности. С увеличением размера пор,
их объём возрастает как
 ,
а площадь как
,
а площадь как ,
за счет этого и наблюдается смещение
максимума дифференциальной кривой.
,
за счет этого и наблюдается смещение
максимума дифференциальной кривой.
Адсорбция на границе твердое тело – жидкость.
В случае адсорбции на границе твердое тело – газ, мы пренебрегали одним компонентом. В случае адсорбции на границе твердое тело – жидкость адсорбат вытесняет с поверхности адсорбента молекулы растворителя.
 ,
,
Справедливо уравнение:

 ,
,
N 1 , N 2 – мольные доли растворителя и компонента, N 1 + N 2 = 1, тогда
 ,
=>
,
=>
 ,
тогда- уравнение адсорбции для границы раздела
фаз твердое тело – жидкость.
,
тогда- уравнение адсорбции для границы раздела
фаз твердое тело – жидкость.
Адсорбция (Г) >
0 при
 <
0
<
0
Если
значения для компонента и растворителя сильно
различны, в этом случае зависимостьГ
от N
имеет экстремум при значении N
~ 0,5.
для компонента и растворителя сильно
различны, в этом случае зависимостьГ
от N
имеет экстремум при значении N
~ 0,5.
Е сли
сли имеют
близкие значения, в этом случае возможно
изменение знака адсорбции. ЗависимостьГ
от N
пересекает ось абсцисс
имеют
близкие значения, в этом случае возможно
изменение знака адсорбции. ЗависимостьГ
от N
пересекает ось абсцисс
Точка пересечения функции Г (N ) с осью абсцисс называется адсорбционным азеотропом . Это значит, что два компонента не могут быть разделены на данном адсорбенте.
Уравнение изотермы адсорбции с константой обмена.
При адсорбции на границе твердое тело – жидкость постоянно происходит перераспределение компонентов между поверхностью адсорбента и объемом раствора.
 -
компоненты
(- - относятся к поверхности)
-
компоненты
(- - относятся к поверхности)

 ,
,
 ,
, .
.
 ,
,


Адсорбция на границе жидкость-газ
Р ассмотрим
изменение концентрационного профиля
по мере пересечения границы раздела
жидкость-газ. Пусть компонент 2 летуч.
ассмотрим
изменение концентрационного профиля
по мере пересечения границы раздела
жидкость-газ. Пусть компонент 2 летуч.
Cs – концентрация в поверхностном слое.
Исходя из определения избыточной адсорбции

Если компонент не летуч, то величина адсорбции запишется следующим образом:
П ри
ри

В уравнении
 природа вещества описывается производной
природа вещества описывается производной .
.
Изотерма поверхностного натяжения может иметь вид 1 или 2:
1 – поверхностноинактивные вещества
2 – поверхностноактивные вещества
Поверхностной активностью g называется способность веществ снижать поверхностное натяжение в системе.

 - толщина
поверхностного слоя
- толщина
поверхностного слоя
C s – концентрация компонента в поверхностном слое
С – объемная концентрация
Для гомологического ряда существует правило:
 - правило Траубо
Дюкло
- правило Траубо
Дюкло
Для гомологического ряда изотерма адсорбции выглядит таким образом:
Вместо A пишем Г, так как адсорбция избыточная в поверхностном слое.
Изотерма поверхностного натяжения:

 - поверхностное
натяжение чистого растворителя.
- поверхностное
натяжение чистого растворителя.
 - фундаментальное
адсорбционное уравнение;
- фундаментальное
адсорбционное уравнение;
 -
уравнение Лангмюра.
-
уравнение Лангмюра.
Решим их совместно:

- уравнение Шишковского.
B – константа для гомологического ряда.
A
- при переходе от одного гомолога к
другому увеличивается в 3-3,5 раза

![]()
1 – область малых концентраций
![]()
2 – средняя концентрация
3 – мономолекулярный слой
Поверхностноактивные вещества представляют собой дифильные молекулы, т.е. включают полярную группу и неполярный углеводородный радикал.
o - полярная часть молекулы.
| - неполярная часть молекулы.
В полярном растворителе молекулы ПАВ ориентируются таким образом, что полярная часть молекулы обращена к растворителю, а неполярная выталкивается в газовую фазу.
В уравнении
Шишковского
 ,
она постоянна для гомологического ряда.
,
она постоянна для гомологического ряда.
Поверхностноактивное действие начинает проявляться с n >5. При концентрациях больших, чем концентрация мономолекулярного слоя, в растворах ПАВ происходит мицеллообразоваие.
Мицелла – называется агрегат молекул дифильных ПАВ, углеводородные радикалы которых образуют ядро, а полярные группы обращены в водную фазу.
Масса мицеллы – мицелляльная масса.
Ч исло
молекул – число агрегации.
исло
молекул – число агрегации.
Сферические мицеллы
В случае мицеллообразования в растворе устанавливается равновесие
ККМ – критическая концентрация мицеллообразования.
Поскольку мы считаем мицеллу отдельной фазой:
Для гомологического ряда существует эмпирическое уравнение:
a – энергия растворения функциональной группы.
b
 – инкремент адсорбционного потенциала,
работа адсорбции на одно метиленовое
звено.
– инкремент адсорбционного потенциала,
работа адсорбции на одно метиленовое
звено.
Наличие в мицеллах углеводородного ядра создает возможность для растворения в водных растворах ПАВ соединений, которые не растворимы в воде, это явление называется солюбилизацией (то, что растворяется – солюбилизат, ПАВ – солюбилизатор).
Грязь может быть совсем не полярна, может содержать как полярную, так и не полярную часть и будет ориентироваться как молекула ПАВ.
В любом случае при солюбилизации происходит увеличение мицеллярной массы и числа агрегации не только за счет включения солюбилизата, но и за счет увеличения числа молекул ПАВ, необходимых для поддержания равновесного состояния.
Солюбилизация тем эффективнее, чем меньше молекулярная масса солюбилизата.

 ~
72 мН\м.
~
72 мН\м.
 ~
33 мН\м.
~
33 мН\м.
Эффективность ПАВ зависит от величины ККМ.
Двухмерное давление поверхностного слоя

→ -силы поверхностного натяжения.
- двухмерное давление.
Поверхностный слой – это сила равная разности поверхностных натяжений раствора ПАВ и чистого растворителя, направленная в сторону чистой поверхности.
Между раствором и поверхностным слоем устанавливается равновесие



При
 существует область, где
существует область, где линейно зависит
от концентрации.
линейно зависит
от концентрации.
Г [моль/м 2 ].
 -площадь, занимаемая
одним молем вещества
-площадь, занимаемая
одним молем вещества
Тогда изотерма
двухмерного давления будет иметь вид

 - изотерма двухмерного
давления.
- изотерма двухмерного
давления.
Зависимость
 отS М:
отS М:

При
 - двухмерное давление резко возрастает.
При
- двухмерное давление резко возрастает.
При двухмерный деформируется, что вызывает
резкий рост
двухмерный деформируется, что вызывает
резкий рост .
.
Пленка с обеих сторон ограниченная одинаковыми фазами называется двусторонней. В таких пленках наблюдается постоянное движение маточного раствора.
Пленки толщиной меньше 5 нм называются черными пленками.

Адсорбционные слои должны обладать двумя характеристиками: вязкость и легкоподвижность, текучесть и упругость.
Эффект Марангони – это самозалечивание.
Треугольник Гиббса,
 - избыточное давление.
- избыточное давление.
Пленка растянулась и за счет того, что часть жидкости ушла, ПАВ усремляются в свободное место. Треугольник Гиббса.
Эффект адсорбционной прочности тел.
На поверхности пленки всегда существует адсорбционный слой, для которого , тогда
Уравнение Лангмюра:


 в двухмерное
давление
в двухмерное
давление
 - аналог уравнения
Шишковского
- аналог уравнения
Шишковского
Электрокинетические явления. Двойной электрический слой (ДЭС).
Модель Гелемгольца. Теория Гуи-Чапмена.
1808 г. Рейс

U – образная трубка, погружают в неё 2 электрода. Закон сообщающихся сосудов нарушается и происходит изменение уровня жидкости в трубке – электрокинетические явления.
Кинетические явления:
Электрофорез
Электроосмос
Потенциал течения (протекания)
Потенциал седиментации
1 и 2 возникают при наложении разности потенциалов, 3 и 4 продавливание и седиментация коллоидных частиц вызывают появление разности потенциалов.
Электроосмосом называется движение дисперсионной среды относительно неподвижной дисперсной фазы под действием электрического тока.
Электрофорез – это движение частиц дисперсной фазы относительно неподвижной дисперсионной среды под действием электрического тока.
П ричина
возникновения электрокинетических
явлений – это пространственного
разделение зарядов и возникновение
двойного электрического слоя.
ричина
возникновения электрокинетических
явлений – это пространственного
разделение зарядов и возникновение
двойного электрического слоя.
Двойной электрический
слой представляет собой плоский
конденсатор, одна обкладка образована
потенциалоределяющими ионами, другая
– противоиноами. Ионы заражены также
как потенциалопределяющие ко-ионы
оттеснены в объем раствора. Расстояние
между обкладками
 .
Потенциал падает линейно, разность
потенциалов
.
Потенциал падает линейно, разность
потенциалов .
.
Внешняя разность
потенциалов вызывает появление модуля
сдвига
 - это пара сил отнесенных к единице
площади, девствующих вдоль поверхности
твердого тела.
- это пара сил отнесенных к единице
площади, девствующих вдоль поверхности
твердого тела.

В состоянии
равновесия модуль сдвига равен модулю
вязкого трения ( ).
).

В наших условиях
 ,
,

 - уравнение
Гелемгольца-Смалуковского
- уравнение
Гелемгольца-Смалуковского
 - линейная скорость
смещении я фаз.
- линейная скорость
смещении я фаз.
E – напряженность электрического поля.
 - разность потенциалов
между обкладками
- разность потенциалов
между обкладками
 - электрофоретическая
подвижность [м 2 /(В*с)].
- электрофоретическая
подвижность [м 2 /(В*с)].
В модели Гелемгольца не учитывается тепловое движение молекул. Реально распределение ионов в двойном слое носит более сложный характер.
Гуи и Чапман выделили следующие причины возникновения ДЭС:
Переход иона из одной фазы в другую при установлении равновесия.
Ионизация вещества твердой фазы.
Достройка поверхности ионами, присутствующими в дисперсионной среде.
Поляризация от внешнего источника тока.
Двойной электрической слой имеет размытое или диффузное строение. Ионы стремятся равномерно распределиться во всем диффузном слое.
Диффузный слой состоит из противоинонв, протяженность слоя определяется их кинетической энергией. При температуре стремящейся к абсолютному нулю противоиноы максимально приближены к потенциалопределяющим ионам.
Даня теория базируется на двух уравнениях:
уравнение Больцмана

 - работа против
сил электростатического взаимодействия.
- работа против
сил электростатического взаимодействия.
 - объёмная плотность
заряда.
- объёмная плотность
заряда.

Уравнение Пуассона

Поскольку толщина
ДЭС много меньше размеров частицы и для
плоского ДЭС производная по координатам
 и
и упраздняется.
упраздняется.

Для е у при у<<1 функцию можно разложить в ряд Маклорена:

Ограничимся двумя членами ряда, тогда:


 -
толщина ДЭС – это расстояние, на котором
потенциал ДЭС уменьшается в e
раз.
-
толщина ДЭС – это расстояние, на котором
потенциал ДЭС уменьшается в e
раз.
Чем меньше
температура, тем меньше
 .
При Т→0 – плоский ДЭС. Чем больше
концентрация, тем большеI,
тем меньше
.
При Т→0 – плоский ДЭС. Чем больше
концентрация, тем большеI,
тем меньше
 .
.





 «–» означает, что
потенциал с расстоянием уменьшается.
=>
«–» означает, что
потенциал с расстоянием уменьшается.
=>
=>

 ,
,
 - потенциал экспоненциально уменьшается.
- потенциал экспоненциально уменьшается.
Потенциал для
поверхностной плотности заряда:

Поверхностный заряд – объемный заряд с противоположным знаком, проинтегрированный по расстоянию.



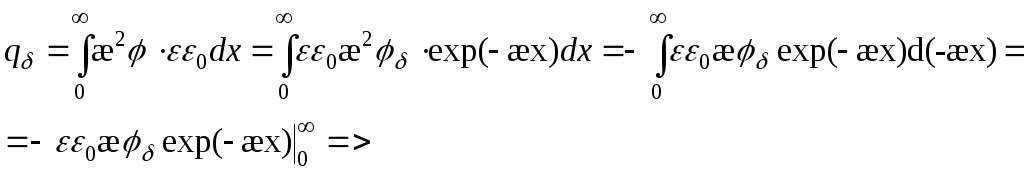
=>


Там, где потенциал
уменьшается в 2,7 раза -

Ёмкость двойного
слоя

Недостаток теории
– не учитывается наличие слоя Гелемгольца,
т.е. не учитывает
 ,
отсюда ошибки в определении основных
параметров. Также не объясняет влияние
ионов различной природы на толщину
двойного электрического слоя.
,
отсюда ошибки в определении основных
параметров. Также не объясняет влияние
ионов различной природы на толщину
двойного электрического слоя.
Теория Штерна. Строение коллоидной мицеллы.
Двойной электрический
слой состоит из двух частей: плотной и
диффузной. Плотный слой образуется в
результате взаимодействия потенциалобразующих
ионов со специфически адсорбирующимися.
Эти ионы, как правило, частично или
полностью дегидратированы и могут иметь
как одинаковый, так и противоположный
к потенциалопределяющим ионам заряд.
Это зависит от соотношения энергии
электростатического взаимодействия
 и потенциала специфической адсорбции
и потенциала специфической адсорбции .
Ионы плотного слоя закреплены. Другая
часть ионов расположена в диффузном
слое, эти ионы свободны и могут перемещаться
вглубь раствора, т.е. из области большей
концентрации в область меньшей. Общая
плотность заряда складывается из двух
частей.
.
Ионы плотного слоя закреплены. Другая
часть ионов расположена в диффузном
слое, эти ионы свободны и могут перемещаться
вглубь раствора, т.е. из области большей
концентрации в область меньшей. Общая
плотность заряда складывается из двух
частей.

 -заряд слоя
Гельмгольца
-заряд слоя
Гельмгольца
 -Заряд диффузного
слоя
-Заряд диффузного
слоя
Поверхность имеет определенное число адсорбционных центров, каждый из которых взаимодействует с одним противоионом. Константа такой квазихимической реакции равна:

 ,
где
,
где
 - мольная доля противоионов в растворе
- мольная доля противоионов в растворе
Распределение Гельмгольца
Потенциал убывает линейно
Распределение
потенциала по Гуи
.
Плотного слоя нет, потенциал убывает
экспоненциально со значения

Распределение по Штерну .
Вначале снижение потенциала линейно, а затем экспоненциально.
При наложении электрического поля в случае электрофореза движется не непосредственно частица твердой фазы, а частица твердой фазы со слоем окружающих её ионов. ДЭС повторяет форму частицы дисперсной фазы. При наложении потенциала отрывается часть диффузного слоя. Линия разрыва называется границей скольжения .
Потенциал,
возникающий на границе скольжения в
результате отрыва части диффузного
слоя, называется электрокинетическим
потенциалом
(Дзэта потенциал
 ).
).
Частица дисперсной фазы, с окружающим её слоем противоионов и двойным электрическим слоем называется мицеллой .
Правила написания коллоидных мицелл:


1-1 зарядный электролит
T – частица дисперсной фазы.
AA – граница плотной и диффузной части.
BB – граница скольжения.
Граница скольжения может совпадать с линией AA, а может и не совпадать.
Значения pH, при котором дзэта-потенциал равен нулю, называется изоэлектрической точкой .
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. В избытке CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
{CaSO 4 m∙nCa 2+ 2(n - x )Cl - } 2 x + x Cl - - запись мицеллы.
CaSO 4 m – агрегат.
CaSO 4 m∙nCa 2+ – ядро.
CaSO 4 m∙nCa 2+ 2(n - x )Cl - - частица.
2. В избытке Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
{CaSO 4 m∙nSO 4 2- 2(n-x)Na + } 2x- 2xNa + - мицелла
CaSO 4 m – агрегат.
CaSO 4 m∙nSO 4 2 + – ядро.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - частица
Уравнение Гелемгольца-Смолуховского

 - линейная скорость
смещения границ (в электроосмосе).
- линейная скорость
смещения границ (в электроосмосе).
 - разность потенциалов
на обкладках конденсатора (в электроосмосе).
- разность потенциалов
на обкладках конденсатора (в электроосмосе).


 - объемная скорость
течения раствора, S
– площадь поперечного сечения ячейки.
- объемная скорость
течения раствора, S
– площадь поперечного сечения ячейки.

E – напряженность электрического поля.


 (для электроосмоса).
(для электроосмоса).
Для потенциала течения:

 - потенциал
- потенциал
 - давление на
мембрану
- давление на
мембрану
Как правило, значение электрофоретических подвижностей и электроосмотических подвижностей меньше расчетных. Это происходит вследствие:
Релаксационного эффекта (при движении частицы дисперсной фазы нарушается симметрия ионной атмосферы).
Электрофоретического торможения (возникновение дополнительного трения в результате движения противоионов).
Искажение линий тока в случае электропроводных частиц.
Связь поверхностного натяжения с потенциалом. Уравнение Липпмана.
Образование ДЭС происходит самопроизвольно вследствие стремления системы снизить свою поверхностную энергию. В условиях постоянства T и p обобщенное уравнение первого и второго законов термодинамики выглядит:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1-е уравнение
Липпмана.
- 1-е уравнение
Липпмана.
 - поверхностная
плотность заряда.
- поверхностная
плотность заряда.
 - дифференциальная
емкость.
- дифференциальная
емкость.
 - 2-е уравнение
Липпмана.
- 2-е уравнение
Липпмана.
С – емкость.
Решим 1-е уравнение Липпмана и фундаментальное уравнение адсорбции:
 ,
,


 ,
тогда
,
тогда
 - уравнение
Нернста
- уравнение
Нернста
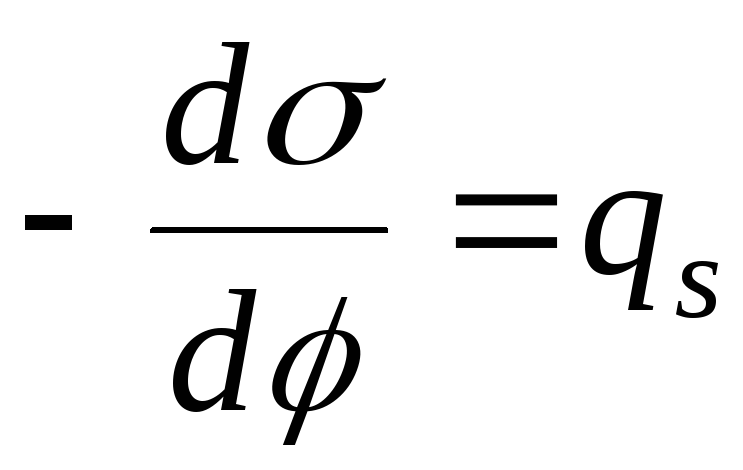 ,
,
 ,
,




 - уравнение
электрокапиллярной кривой (ЭКК).
- уравнение
электрокапиллярной кривой (ЭКК).
В
 :
: ,
но
,
но
Катионные ПАВы (КПАВ) снижают катодную ветвь ЭКК.
Анионные ПАВы (АПАВ) снижают анодную ветвь ЭКК.
Неионогенные ПАВы (НПАВ) снижают среднюю часть ЭКК.
Устойчивость дисперсных систем. Расклинивающее давление.
Дисперсные системы можно разделить:
Системы неустойчивые термодинамически могут быть устойчивы кинетически за счет перехода в метастабильное состояние.
Различают два вида устойчивости:
Седиментационная устойчивость (по отношению к силе тяжести).
Агрегативная устойчивость. (по отношению к слипанию)
Коагуляция – это процесс слипания частиц, приводящий к потере агрегативной устойчивости. Коагуляцию может вызвать изменение температуры, pH, перемешивание, ультразвук.
Различают коагуляцию:
Обратимая.
Необратимая.
Коагуляция протекает при введении электролитов.
Правила коагуляции:

Плёнка – это часть системы, находящаяся между двумя межфазными поверхностями.
Расклинивающее давление возникает при резком уменьшении толщины плёнки в результате взаимодействия сближающихся поверхностных слоев.

«-» - при уменьшении толщины пленки расклинивающее давление возрастает.
P 0 – давление в объемной фазе, которая является продолжением прослойки.
P 1 – давление в пленке.
Теория устойчивости. ДЛФО (Дерягин, Ландау, Фервей, Овербек).
Согласно теории ДЛФО в расклинивающем давлении выделяют две составляющие:
Электростатическая П Э (положительная, она обусловлена силами электростатического отталкивания). Соответствует уменьшению энергии Гиббса при возрастании толщины пленки.
Молекулярная П М (отрицательная, обусловлена действием сил притяжения). Обусловлена сжатием пленки за счет химических поверхностных сил, радиус действия сил десятые доли нм с энергией порядка 400 кДж/моль.
Полная энергия
взаимодействия
:

 - система агрегативно
устойчивая
- система агрегативно
устойчивая
 - неустойчивая
система
- неустойчивая
система
П оложительная
составляющая.
оложительная
составляющая.
Увеличение обусловлено увеличением потенциальной энергии при сжатии тонких пленок. Для пленок большой толщины избыточная энергия ионов скомпенсирована и равна энергетическому взаимодействию в объеме дисперсионной среды.
Если
 (
( - толщина пленки,
- толщина пленки, -
радиус иона) утоньшение пленки приводит
к исчезновению и уменьшению в ней молекул
и ионов с минимальной поверхностной
энергией. Число соседних частиц
уменьшается, в результате чего
потенциальная энергия оставшихся в
пленке частиц возрастает.
-
радиус иона) утоньшение пленки приводит
к исчезновению и уменьшению в ней молекул
и ионов с минимальной поверхностной
энергией. Число соседних частиц
уменьшается, в результате чего
потенциальная энергия оставшихся в
пленке частиц возрастает.


Теория ДЛФО взаимодействие частиц рассматривает как взаимодействие пластин.

Частицы не взаимодействуют



 - уравнение Лапласа,
- уравнение Лапласа,
 ,
,


Для слабо заряженных поверхностей
Для сильно заряженных поверхностей:

Молекулярная составляющая – взаимодействие двух атомов:
 ~
~
Взаимодействие атома с поверхностью:

Возьмем две пластинки:
Д ля
получения молекулярной составляющей
необходимо провести суммирование всех
энергий взаимодействия атомов правой
и левой пластин.
ля
получения молекулярной составляющей
необходимо провести суммирование всех
энергий взаимодействия атомов правой
и левой пластин.
где
 - постоянная Гамакера (учитывает природу
взаимодействующих тел).
- постоянная Гамакера (учитывает природу
взаимодействующих тел).
Т.о. энергия взаимодействия частиц в системе может быть выражена с помощью потенциальных кривых.
I – первичный потенциальный минимум. Это зона необратимой коагуляции, силы притяжения преобладают.
II – зона агрегативной устойчивости, преобладают силы отталкивания.
III – вторичный потенциальный минимум (или зона флокуляции). Между частицами дисперсной фазы существует прослойка электролита, и частицы могут быть разделены и переведены в зону агрегативной устойчивости.

Кривая 1 – система агрегативно устойчива.
Кривая 2 – в зоне I устойчива, в зоне II не устойчива.
Кривая 3 – в системе произошла коагуляция.
Кривая 4 – в точке
4 суммарная энергия взаимодействия U=0,
 ,
эта точка экстремума соответствует
началу быстрой коагуляции.
,
эта точка экстремума соответствует
началу быстрой коагуляции.
Существует два случая:
1. Поверхности слабозаряженные:
U = U Э + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - это толщина
прослойки соответствующая началу
процессу коагуляции.
- это толщина
прослойки соответствующая началу
процессу коагуляции.






 - для слабозаряженных
поверхностей
- для слабозаряженных
поверхностей
 тогда
тогда

2. Для сильнозаряженных поверхностей:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Возведем (3) в квадрат

Коагуляция:
При специфической адсорбции ионы могут адсорбироваться в сверхэквивалентном количестве таким образом, что поверхность может изменить свой заряд. Происходит перезарядка поверхности.
В случае специфической адсорбции могут адсорбироваться ионы не только противоположных знаков, но и одного.
Если адсорбируются ионы того же знака, что и поверхность, то в поверхностном слое будет происходить не падение потенциала, а его рост.

Нейтрализационная коагуляция (протекает с участием слабозаряженных частиц и зависит не только от заряда электролита-коагулятора, но и от потенциала на границе плотного и диффузного слоя).
Теория быстрой коагуляции Смолуховского.

Зависимость скорости коагуляции от концентрации электролита.
I – скорость коагуляции мала,
II – скорость коагуляции практически пропорциональна концентрации электролита.
III – область быстрой коагуляции, скорость практически не зависит от концентрации.
Основные положения :
Исходный золь монодисперсный, сходные частицы имеют сферическую форму.
Все столкновения частиц результативны.
При столкновении двух первичных частиц образуется вторичная. Вторичная + первичная = третичная. Первичное, вторичное, третичное – кратность.
В терминах химической кинетики процесс коагуляции может быть описан уравнением:

Решением будет
уравнение:

 - время половинной
коагуляции. Это время, в течение которого
число частиц золя уменьшается в 2 раза.
- время половинной
коагуляции. Это время, в течение которого
число частиц золя уменьшается в 2 раза.

 ,
,
 ,
,
 ,
,


По мере увеличения
кратности максимум кривых коагуляции
сдвигается в сторону больших значений
 .
.
Недостатки:
Предположение о монодисперсности.
Предположение о результативности всех столкновений.